Shagun Aggarwal,MD OBG,DM Medical Genetics
Additional Professor, Department of Medical Genetics, Nizam’s Institute of Medical Sciences, Hyderabad
Adjunct Scientist, Diagnostics Division, Centre for DNA Fingerprinting & Diagnostics, Hyderabad.
email:shagun.genetics@gmail.com
Introduction
Skeletal dysplasias are a group of >400 genetic diseases affecting the developing chondro-osseous tissue and resulting in short stature, bony deformities and other co-morbidities.Of these, at least 100 disorders can present prenatally, usually in the form of short bones—detailed ultrasonography ascertainsessential prognostic and diagnostic information, hence, guiding pregnancy management decisions.A genetic evaluation is an integral component of this diagnostic odyssey. Knowledge of the various skeletal dysplasia, their gestational age of onset, specific ultrasound findings and underlying genetic and mutational spectrum is pivotal to establishing a final diagnosis.This writeup highlights the various skeletal dysplasiaspresenting to a imaging specialist, the diagnostic approach from a clinical genetic perspective and the relevant laboratory testing modalities.
The magnitude of skeletal dysplasias as a perinatal problem
Skeletal dysplasias(SKDs) are reported to have a birth prevalence of 2.4-4.6 per 10,000live births.The Nosology of Genetic disorders ofSkeletonn,2019 lists at least 461 different diseases under this large group.Of these, >100 are recognisablein the perinatal period and 50% of these are lethal.Overall, these account for 1-2% of all stillbirths.Some of the commonest SKDs at birth are Achondroplasia seen in 1 in 15000-50000, Thanatophoric dysplasia present in 1 in 17000-50000 and Osteogenesis imperfecta seen in 1 in 10000-20000liveborns.In the prenatal period, assessment of perinatal lethality is the single most important parameter that has implications for important decisions regarding continuation or termination of pregnancy.A list of the Lethal and Non-lethal SKDs presenting in the perinatal period has been provided in table 1.Besides the criteria of lethality, recognition of these individual disorders in the prenatal period using imaging and genetic testing plays an important role ina more accurate postnatal prognostication, and this can help the family make more informed decisions.Also, these disorders have varying genetic basis and inheritance patterns.This has important implications for the genetic tests offered and the recurrence risk counselling for the couple.
Postnatal clinical outcomes of SKDs
Perinatal Lethality
Perinatal lethality is the most important parameter on which important reproductive decisions are based on prenatal detection of a SKD.At least half of prenatally detected SKDs are lethal, the primary cause of this being pulmonary hypoplasia due to a small thoracic cage.Prenatally, this is usually determined on basis of the thoracic circumference and degree of long bone shortening using various biometric measures and ratios.In many common SKDs like Short rib thoracic dysplasias(includes short rib polydactyly syndromes, Jeune’s asphyxiating thoracic dystrophy and Ellis Van Crevald syndrome, Thanatophoric dysplasia and Osteogenesis imperfecta, a small thoracic cage is the primary cause of perinatal death.In addition, other abnormalities like visceral malformations (Short rib thoracic dysplasias) and tracheo-laryngomalacia in Campomelic dysplasia may also result in a lethal outcome.
Short stature
All skeletal dysplasias are associated with short stature in postnatal period.The degree of short stature and final adult height varies amongst the different conditions.However, most prenatally detected disorders have significant short stature with adult heights being below 135-140cm.Some diseases where the degree of short stature may be relatively milder are Jeune’s thoracic dystrophy,X-linked chondrodysplasia punctata, Campomelic dysplasia and some syndromic forms like Robinow syndrome.
Most skeletal dysplasias result in disproportionate short stature, either with abnormally short limbs or abnormally short trunk or a combination of both.In addition, there could be various deformities like bowing of long bones (Achondroplasia, Campomelic dysplasia), spinal and chest deformities(Spondyloepiphyseal dysplasias, Jeune’sthoracic dystrophy) or disabling,recurrent fractures(Osteogenesis imperfecta, Hypophosphatasia).These result in significant cosmetic and functional handicaps for the affected individuals.
Other co-morbidities of SKDs
Certain SKDs have associated visceral malformations e.g. polydactyly, renal and cardiac abnormalities in the various Short rib thoracic dysplasias, ambiguous genitalia in Campomelic dysplasia, cleft palate in Diastrophic dysplasia, Ateleosteogenesis and Oto-palatodigital syndrome.In addition, some SKDs can have functional deficiencies not detectable on ultrasound imaging, e.g. intellectual disability in some patients with Jeune’s thoracic dystrophy, Rhizomelic chondrodysplasia punctata and other syndromic forms like Oro-facial digital syndrome and Oto-palatodigital syndrome.Hearing deficits can be seen in patients with Fibrochondrogenesis, Oto-palatodigital syndrome and others.
In view of these multisystemic involvement in many of the skeletal dysplasias, the prenatal establishment of a specific genetic diagnosis is important for accurate for postnatal prognostication, even in apparently non-lethal disorders.Such a diagnoses can also help in anticipatory guidance for neonatal management and subsequent postnatal care.
Prenatal imaging presentations of Skeletal dysplasias
A prenatal onset skeletal dysplasia can present with one or more of the following ultrasound findings:i) Femur length < 2SD for gestation, ii) Increased nuchal translucency, iii) Hydrops, iv) Increased visibility of intracranial structures, v) Abnormal bones- bent, fractures, vi) Small thorax, abnormal skull shape
1.Short femur: This is the most common ultrasound finding that raises suspicion of a SKD.The degree of femur shortening has important implications on the likely differential diagnoses.Hence, it is important to chart the femur length on percentile charts and calculate the exact Z scores.A similar exercise should be done for the other long bones, as this gives an idea regarding the pattern of limb segment involvement, eg.rhizomelia(proximal limb segment shortening), mesomelia(middle segments shortening) and this further helps in reaching a specific genetic diagnosis.
If femur length is between -2 to -4SD for the gestation, besides a SKD the following differentials are also possible:i) Fetal growth restriction, ii) Chromosomal disorders, iii)Monogenic syndromes
It is important to assess fetus for other malformations and facial dysmorphism which may indicate possibility of a chromosomal or monogenic syndrome; and also to look for evidence of fetal growth restriction in order to distinguish these alternative causes of a short femur.
Some of the skeletal dysplasias which present with such milder degree of shortening are discussed below.Most of these disorders become apparent by the later part of the second trimester of pregnancy.
- a) Achondroplasia: This is one of the most common skeletal dysplasia seen at birth.In prenatal period, it usually presents with mild degree of long bone shortening starting at 24 weeks of gestation.This is an autosomal dominant disorder with mutations in the FGFR3 gene.Most cases occur due to de-novo mutations in a healthy parents, but in some families it may be inherited from an affected parent. >95% patients have a common mutation, c.1138 G>A, and hence genetic testing can be done easily by targeted mutation analysis in a basic molecular genetic laboratory in a timely and cost-effective manner.This is a nonlethal condition, and the primary postnatal problem is the short stature.
b) Jeune’s Asphyxiating Thoracic dystrophy: This is an autosomal recessive disorder which is a type of short rib thoracic dysplasia.These fetuses present with a narrow, bell shaped thorax, short ribs and long bone shortening which is primarily rhizomelic.Additional malformations can be present like polydactyly, hepatic and renal abnormalities in some patients.Around 40% of these fetuses will be perinatal lethal, while those surviving into postnatal period may develop renal or hepatic failure.This is a genetically heterogenous disorder and genetic testing requires next generation sequencing based exome sequencing or multi gene panel testing.Recurrence risk in subsequent conceptions is 25%.
c) Ellis Van crevald syndrome: This is also a type of short rib thoracic dysplasia which has additional multisystemic involvement in form of ectodermal dysplasia, polydactyly and cardiac defects.The long bone shortening involves the middle and distal segments (acro-mesomelic) and narrow, cylindrical thorax with short ribs can be appreciated on imaging.Inheritance is autososmal recessive with mutations in EVC1 or EVC2 genes, testing can be done by Sanger sequencing or next generation sequencing based tests.Many of these babies would survive the perinatal period.
d)X-linked chondrodysplasia punctata: This is a rarer skeletal dysplasia with mild degree of bone shortening.The most striking feature is Binder facies on ultrasound imaging which can be seen in early second trimester.Subsequent prognosis is favorable with mild short stature, no pulmonary hypoplasia or systemic involvement.
If femur length is < -4SD , the most likely diagnosis is a skeletal dysplasia, possibly a lethal one.Other differentials in such a scenario could be conditions with limb reduction defects eg.Focal femoral hypoplasia, Robert’s syndrome, phocomelia, etc.Most of the skeletal dysplasia with such a degree of shortening present in first or early second trimester.Some of the conditions in this category are as follows.
a)Thanatophoric dysplasia:This is a lethal SKD caused by de-novo mutations in FGFR3 gene.It manifests in first or early second trimester with very short limbs, short narrow thorax , a relatively large head and associated hydrops or increased NT.Two distinct phenotypes arerecognised, type I with bent femurs(telephone receiver shaped) and normal shape of head, and type II with clover leaf shaped skull and normal contour of femur.Ventriculomegaly, migrational abnormalities of brain and rarely cardiac, renal defects are also reported.Prognosis is poor, however recurrence risk is low.
- b) Osteogenesis imperfecta type II: : A lethal skeletal dysplasia which is characterisedby poor skeletal mineralisation ,multiple fractures and bending of long bones and ribs.It may present in first or second trimester of pregnancy.Unique features are increasedcompressibility of skull, improved visibility of intracranial structures and acute angulation of tubular bones.Majority of cases are due to de-novo mutations in COL1A1 and COL1A2 genes, however many autosomal recessive forms with mutations in various genes are also known.Genetic testing is using Next generation sequencing based exome sequencing or multigene panel testing.
c)Short rib thoracic dysplasias: These are a group of genetically heterogenous skeletal dysplasiasdue to mutations in various genes involved in primary cilia formation and function.all characterised by extremely short ribs and narrow thorax, with or without polydactyly and other visceral malformations.Presentation may be in first trimester or early second trimester.Genetic testing is using Next generation sequencing based exome sequencing or multigene panel.
d) Achondrogenesis: This is a skeletal dysplasia with extremely short long bones, relatively large head and ossification abnormalities of the skull, spine and pelvis.Three types characterised, type 1a,1b and type 2, these being caused by mutations in TRIP11, SLCA26A2 and COL2A1 genes respectively.inheritance is autosomal recessive in type 1a & 1b, and autosomal dominant due to de-novo mutations in type 2.Due to extreme limb shortening, these are usually reconsidered in first trimester ultrasound, and often have associated hydrops.Genetic testing is using Next generation sequencing based exome sequencing or multigene panel.
2.Bent bones: Certain SKDs present with bent bones on ultrasound, most often along with concomitant shortening.Of these, Osteogenesis imperfecta and thanatophoric dysplasia are themost common types.In addition, certain other SKDs also have bent bones as a prominent feature.Campomelic dysplasia is one such SKD that usually has mild bending of bilateral femurs.Degree of shortening is not significant in this condition, usually between -2 to -4SD.The most commonly involved bones are femur, humerus and tibia.In addition, some unique findings can be appreciated, in form of hypoplastic scapula, 11 pair of ribs, micrognathia and sex reversal/genital ambiguity in male fetuses.It is caused by mutations in SOX9 genes, and is an autosomal dominant condition.Perinatal lethality may occur in few cases due to laryngomalacia.However, most patients would survive into postnatal period with short stature and long bone bending of varying degrees.
3.Hydrops or increased NT: Some of the lethal skeletal dysplasias can present with increased NT or grossly hydropic appearance.These include achondrogenesis, atelosteogenesis, short rib thoracic dysplasias, osteogenesis imperfecta and thanatophoric dysplasia.
4.Syndromic disorders with prominent skeletal involvement: In certain genetic syndromes, skeletal involvement with shortening/other abnormalities of bones is a predominant finding.These are also grouped under genetic disorders of theSkeleton as per the Nosology, 2019.These mimic the skeletal dysplasia on antenatal ultrasound, and can be differentiated based on the additional findings and./or genetic testing
a)Robinow syndrome: This is a condition with prominent mesomelic shortening of long bones, more significantly involving the upper limbs.In addition, other characteristic features can be appreciated in form of vertebral segmentation defects and facial dysmorphism.Postnatal outcome is fair, with short stature being the primary morbidity in most cases.
- b)Oro-facial digital syndrome: These are a group of conditions characterised by polydactyly, variable oro-facial clefts, tongue lobulation or hamartomas with or without malformations involving brain and kidneys.They overlap with the short rib thoracic dysplasias, and may not be differentiated on antenatal ultrasound without genetic testing.postnatal prognosis is usually guarded due to multisystemic involvement.
- c)Oto-palato-digital syndrome(OPD): OPD 1 & 2 are X-linked disorders caused by mutations in the FLNA genes.They are characterised by micrognathia, cleft palate, hand and feet deformities, with or without visceral malformations.Postnatal prognosis is guarded, especially in males
- d)Microcephalic osteodysplastic dwarfism: These are a group of disorders usually associated with symmetric bone shortening, overall fetal growth restriction and prominent microcephaly.Postnatal prognosis is variable as per the individual disorder, however usually extreme short stature and microcephaly are present.
Approach to ultrasound for genetic diagnoses
Antenatal ultrasound can be used to arrive at a presumptive genetic diagnoses of a specific SKD by paying attention to various findings as mentioned in table 2.Briefly, besides assessment of lethal/non lethal condition, assessment of degree of shortening, pattern of limb segment involvement ,mineralisation, associated malformations, timing of diagnoses and other findings as discussed in individual conditions above; can help in suspecting a specific SKD.This can further be used to counsel families and perform the specific genetic investigation.Consultation with a Clinical geneticist should be sought for pattern recognition and genetic differential diagnoses, and for decision regarding the best laboratory testing approach ,individualised for the patient.
Role of Postmortem examination
A definitive ultrasound diagnosis is possible in only 35-68% cases.Postmortem examination of the fetus including external dysmorphology, radiographs, internal dissection and tissue histopathology is very important for detailed phenotype delineation.This evaluation should be done in a multidisciplinary manner involving a clinical geneticists and a perinatal pathologist.The post-mortem findings also help in reverse phenotyping and geneotype-phenotype correlation after the genetic test results are available.
Genetic testing for Skeletal dysplasias
SKDs are genetically heterogeneous, and mutations in more than 450 different genes can present with abnormalities of theSkeletonAll these are Mendelian/Monogenic diseases, and cannot be diagnosed by cytogenetic tests like karyotype or chromosomal microarray.Few of the SKDs like Achondroplasia are caused by one specific mutation in majority of patients, and this can be diagnosed using a simple Polymerase chain reaction in a targeted manner.Similarly, campomelic dysplasia and Ellis Van crevald diseases are genetically homogenous and targeted gene sequencing can be done for these using conventional Sanger sequencing.However, for most other SKDs, genetic heterogeneity is the norm, and hence the best diagnostic approach is to use a Next generation sequencing based Exome sequencing or Skeletal dysplasia gene panel.This is especially relevant in the prenatal period, when the specific diagnosis may not be certain.NGS based testing is now available in various laboratory across India at a reasonable cost.The turnaround time ranges from 2-4 weeks, and this may limit its utility in the prenatal period.Also, this testing is likely to reveal variants of uncertain significance and other confusing results like incidental or secondary findings.Hence, this should be performed in collaboration with a Clinical geneticist who should be involved in the Pre- test and post-test counseling of the patients.Besides, confirmation of a specific genetic diagnosis, identification of the causative mutation helps in providing accurate recurrence risks and opportunity for early, definitive prenatal testing or preimplantation genetic testing for these families.
Conclusions
SKDs are a large group of genetic disorders presenting the fetal life with skeletal abnormalities.These disorders have important implications for postnatal outcome both in terms of short stature and various other disabling morbidities, a significant proportion being perinatal lethal.A multidisciplinary approach towards ultrasound diagnosis and laboratory genetic testing with involvement of a clinical geneticist is essential for management of pregnancies affected with a SKD.In view of significant recurrence risk for such families, genetic counselling is an important component of the clinical protocol.
Table 1. List of some common prenatally detected Skeletal dysplasias
| Lethal Skeletal Dysplasias |
Gene involved |
| Thanatophoric dysplasia |
FGFR3 |
| Homozygous achondroplasia |
FGFR3 |
| Achondrogenesis |
COL2A1,TRIP11,SLC26A2 |
| Short rib thoracic dysplasias |
WDR60, WDR35,WDR34, TCTEX1D2,SRTD12,NEK1,KIAA0753,KIAA0586,INTU,IFT81,IFT52,IFT43,IFT172,IFT140,DYNC2LI1,CEP120 |
| Campomelic dysplasia |
SOX9 |
| Hypophosphatasia |
ALPL |
| Jeune’s asphyxiating thoracic dystrophy |
DYNC2H1IFT80, WDR19,TTC21B, SRTD1 |
| Atelosteogenesis- I,II,III |
FLNB,SLC26A2 |
| Diastrophic dysplasia |
SLC26A2 |
| Fibrochondrogenesis |
COL11A1,COL11A2 |
| Non Lethal Skeletal Dysplasias |
| Achondroplasia |
FGFR3 |
| Jeune’s asphyxiating thoracic dystrophy |
DYNC2H1IFT80, WDR19,TTC21B, SRTD1 |
| Ellis Van Crevald syndrome |
EVC1,EVC2 |
| Osteogenesis imperfecta II |
COL1A1,COL1A2,CRTAP, P3H1,
SPARC,TENT5A,KDELR2,BMP1,
TMEM38B,IFITM5,CREB3L1,
SERPINH1,WNT1,SP7,PPIB,MESD,
SERPINF1,FKBP10,MBTPS2
COL2A1,TRIP11,SLC26A2
WDR60, WDR35,WDR34, TCTEX1D2,SRTD12,NEK1,
KIAA0753,KIAA0586,INTU,IFT81
IFT52,IFT43,IFT172,IFT140
DYNC2LI1,CEP120
SOX9
ALPL
DYNC2H1IFT80, WDR19,TTC21B
, SRTD1
FLNB,SLC26A2
SLC26A2
COL11A1,COL11A2
Non Lethal Skeletal Dysplasias
FGFR3
DYNC2H1IFT80, WDR19,TTC21B
, SRTD1
EVC1,EVC2
COL1A1,COL1A2,CRTAP, P3H1,
SPARC,TENT5A,KDELR2,BMP1,
TMEM38B,IFITM5,CREB3L1,
SERPINH1,WNT1,SP7,PPIB,MESD,
SERPINF1,FKBP10,MBTPS2 |
| Metatropic dysplasia |
TRPV4 |
| Spondyloepiphyseal dysplasia congenita |
COL2A1 |
| Campomelic dysplasia |
SOX9 |
| Opismodysplasia |
INPPL1 |
| Diastrophic dysplasia |
SLC26A2 |
| Kneist Dysplasia |
COL2A1 |
Table 2: Distinguishing ultrasound findings in some Skeletal dysplasias
| Bent bones |
| Osteogenesis imperfecta |
| Thanatophoric dysplasia type 1 |
| Campomelic dysplasia |
| Kyphomelic dysplasia |
| StuveWeidemann syndrome |
| Poor mineralisationion |
| Osteogenesis imperfecta |
| Hypophosphatasia |
| Achondrogenesis |
| Atelosteogensis |
| Large head |
| Thanatophoric dysplasia |
| Achondrogenesis |
| Achondroplasia |
| Cleft lip/palate |
| Short rib thoracic dysplsias |
| Oto-palatodigital syndrome |
| Oro-facial digital syndrome |
| Atelosteogenesis |
| Diastrophic dysplasia |
| Renal/brain/cardiac malformations |
| Short rib thoracic dysplasias |
| Oto-palatodigital syndrome |
| Oro-facial digital syndrome |
| Hitch Hiker thumb |
| Atelosteogenesis |
| Diastrophic dysplasia |
| Vertebral segmentation defect |
| Robinow syndrome |
Figure:
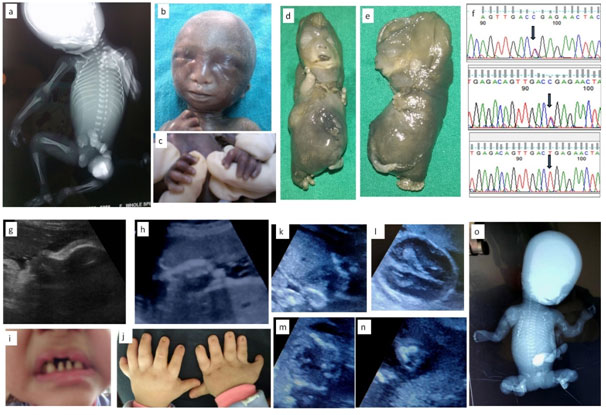
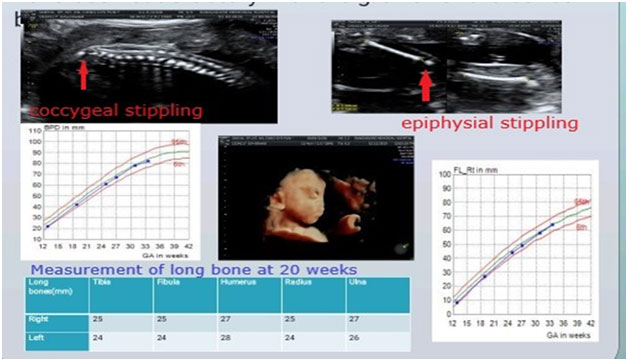
1a-c: Fetus with X linked chondrodysplasia punctata; 1a: Radiograph showing epiphyseal stippling at multiple joints, 1b:Binder facies , 1c: Brachytelephalyngy
1d-f: week Fetus with Achondrogenesis type; 1b,e: Hydrops, extremely short limbs; 1f: Sanger sequences of and parents with c.532C>T, p.Arg178* mutation in exon 2 of SLC26A2 gene
1g-j: Ultrasound and Neonatal photo of fetus with Ellis Van Crevald syndrome; 1g: Depressed nasal bridge and tall forehead on sagittal facial profile, 1h:Postaxial polydactyly , 1i: Dental dysplasia, 1j: Nail dysplasia
1k-o:Fetus with Osteogenesis Imperfecta; 1k: Coronal image of chest showing narrow and bell shaped thorax, 1l: Head showing increasing visibility of intracranial structures, 1m: narrow thorax with short,irregular ribs, 1n: Acutely bent long bone, io: Fetal radiograph showing poor bomineralisationion, short, irregular ribs and short ,irregular long bones with acute angulations